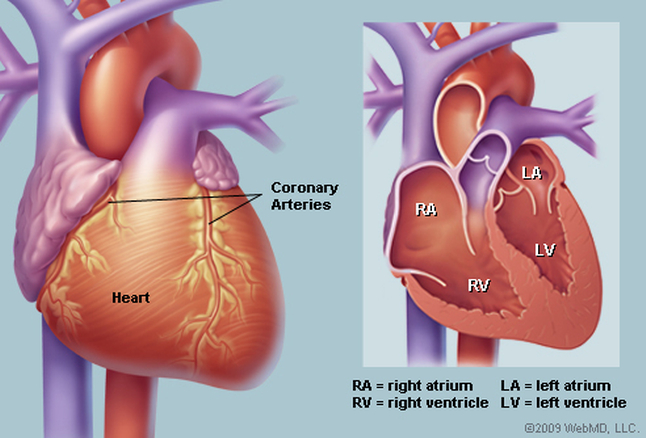
PULMONARY ATRESIA WITH VENTRICULAR SEPTAL DEFECT
Pulmonary atresia with ventricle septal defect, sometimes also called tetralogi of Fallot with pulmonary atresia, shares the intracardiac anatomy of tetralogy but lacks a direct communication between the right ventricle and pulmonary artery. Patients with pulmonary artery and ventricle septal defect have a good size right ventricle, and from this perspective are suitable biventricular repair, major problems with pulmonary artery are common and determine both clinical presentation and management (the complexity of the pulmonary vascular bed may make repair unattractive or impossible).
Early clinical presentation varies between cyanosis (for patients with reduced or threathened pulmonary blood flow), failure to thrive, and/or exertional dyspnoea, to heart failure (for patients with execessive pulmonary blood flow through large major aortic pulmonary collaterals, who may develop segmental pulmonary arterial hypertension with time).
When pulmonary blood flow is is duct dependent, profound cyanosis and cardiovascular collapse ensue as the duct closes. Patients with confluent, good size pulmonary arteries and a pulmonary trunk (usually with valvar atresia) are suitable for a Fallot-like repair using a trans annular patch. Patients with good sized pulmonary arteries but without a pulmonary trunk should undergo repair with a right ventricle-pulmonary arteries conduit. Patients with confluent but hypoplastic pulmonary arteries often require an arterial shunt or reconstruction of the right ventricle outflow tract (without ventricle septal defect closure), which may enhance pulmonary arterial growth, and then be reviewed at a later stage for repair using a valved conduit. Patients with non-confluent pulmonary arteries with adequate, but not excessive, pulmonary blood flow in infancy can survive into adulthood without surgery. There are proponents of a staged unifocalization approach for this latter challenging group of infants, ultimately aiming for a conduit repair.
Late clinical presentation for repaired patients is similar to those with tetralogy, whereas unrepaired patients present with exertional dyspnoea, fatigue and progressive chronic cyanosis, the latter leading to multi organ involvement and, with time, to a number of complications:
- Hemoptysis may be due to rupture of usually small collateral vessels and/or pulmonary artery thrombosis
- Chronic heart failure is usually multi factorial and may be due to chronic cyanosis, early excessive pulmonary blood flow, increased pulmonary vascular resistence, right ventricle dysfunction, aortic regurgitation
- Progressive dilatation of the ascending aorta with increasing aortic regurgitation and aortic disecction (very rare complication) may occur
- Endocarditis can be particularly compromising in patients with limited cardiovascular reserved and those with significant cyanosis
- Increasing cyanosis may be due to decreased pulmonary blood flow from collateral stenosis, pulmonary artery stenosis, increased pulmonary vascular resistance or increasing ventricular end diastolic pressure
- Arrhytmia and sudden cardiac death are not uncommon