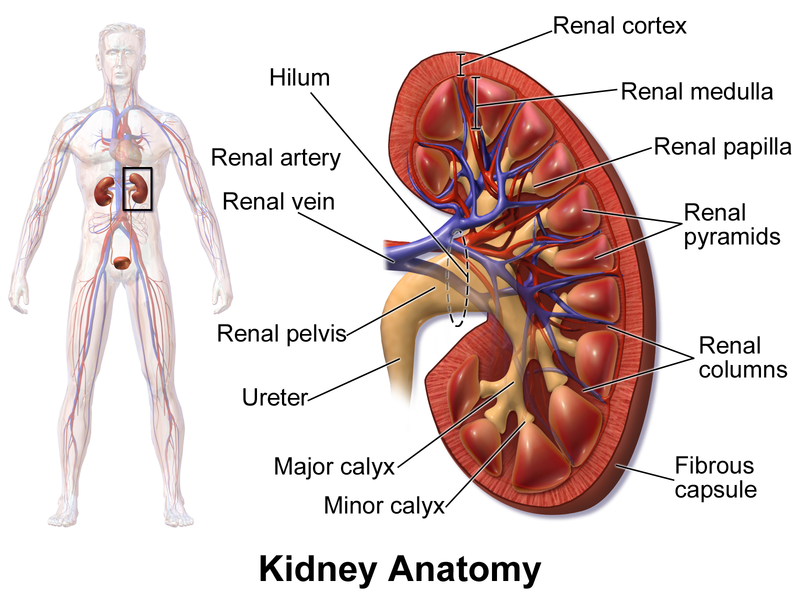
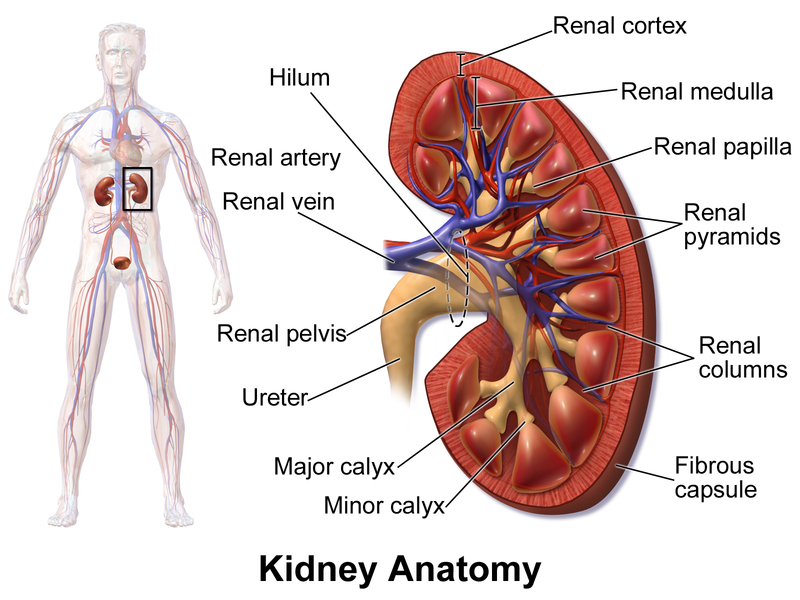
Nephrons are the structural and functional units of the kidneys. Each kidney contains over 1 million of these tiny blood-processing units, which carry out the process that form urine. In addition, there are thousands of collecting ducts, each of which collects fluid from several nephrons and conveys it to the renal pelvis.
Each nephron consist of
a glomerulus, which is a tuft of capillaries, and
a renal tubule. The renal tubule has a cup shaped-end, the glomerular capsule (or Bowman's capsule), which is blind and completely surrounds the glomerulus, much as a well-worn baseball glove encloses a ball. Collectively, the glomerular capsule and the enclosed glomerulus are called
the renal corpuscle.
The endothelium of the glomerular capillaries is fenestrated (penetrated by many pores), which makes them exceptionally porous. This allows large amounts of solute-rich, virtually protein-free fluid to pass from the blood into the glomerular capsule. This plasma-derived fluid or filtrate is the raw material that the renal tubules process to form urine.
The external parietal layer of the glomerular capsule is simple squamous epithelium. This layer simply contributes to the capsule structure and plays no part in forming filtrate.
The visceral layer, which clings to the glomerular capillaries, consist of highly modified, branching epithelial cells called podocyte. The octopus-like podocytes terminate in foot processes, which intertwine as they cling to the basement membrane of the glomerulus. The clefts or opening between the foot processes are called filtration slits. Through these slits, filtrate enters the capsular space inside the glomerular capsule.
The remainder of the renal tubule is about 3 cm long and has three major parts. It leaves the glomerular capsule as the elaborately coiled
proximal convulated tubule, make a hairpin loop called t
he loop of Henle (also called the nephron loop or Henle's loop), and then winds and twists again as
the distal convoluted tubule before emptying into a collecting duct. The terms proximal and distal indicate relationship of the convoluted tubules to the renal corpuscle-filtrate from the renal corpuscle passes through the proximal convulated tubule first and then the distal convulated tubule, which is thus "further away" from the renal corpuscle. The meandering nature of the renal tubule increases its length and enhances its filtrate processing capabilities.
The collecting ducts, each of which receives from may nephrons, run through the medullary pyramids and give them their striped appearance. As the collecting ducts approach the renal pelvis, they fuse together and deliver urine into the minor calyces via papillae of the pyramids.
The U-shaped loop of Henle has decending and ascending limbs. The proximal part of the decending limb is continuous with the proximal tubule and its cells are similar. The rest decending limb, called
the thin segment, is a simple squamous epithelium freely permeable to water. The epithelium becomes cuboidal or even low columnar in the ascending part of the loop of Henle, which therefore becomes
the thick segment. In some nephrons, the thin segment is found only in the descending limb. In others, it extends into the ascending limb as well.
Nephrons are generally divided into two major groups. Cortical nephrons represent 85% of the nephrons in the kidneys. Except for small parts of their loops of Henle that dip into the outer medulla, they are located entirely in the cortex. The remaining
juxtamedullary nephrons originate close to the cortex-medulla junction, and they play an important role in the kidneys ability to produce concentrated urine. Their loops of Henle deeply invade the medulla, and their thin segments are much more extensive than those those of cortical nephrons.